随着欧盟MDR/IVDR法规全面实施,欧洲医疗器械市场的准入壁垒显著提高。MDR法规的申请流程体系复杂、耗时良久,却是企业进军欧洲市场无法绕开的关键环节。为助力企业顺利通过这一挑战,我司于10月15日下午成功举办了第六十二期江苏贸促会展外贸大讲堂活动,特别邀请专家老师,聚焦于MDR申请中两大核心议题――技术文档的编写要点与公告机构的相关要求,为计划或即将启动注册的企业提供极具实用价值的指导。本次会议吸引了来自医疗行业的60多家企业踊跃上线参与。
所有技术文件准备工作都必须严格遵循医疗器械法规(MDR)的框架,这是确保产品合规上市的基本前提。在实际编写技术文档的过程中,企业常常面临诸多挑战:例如验证报告格式或内容不符合规定要求、支撑性数据不充分或缺乏代表性、以及文件体系的齐全性与一致性存有疑问等。这些问题至关重要,直接关系到技术文档评审的通过与否,甚至影响整个申请流程的成败,因此必须从源头上予以高度重视。
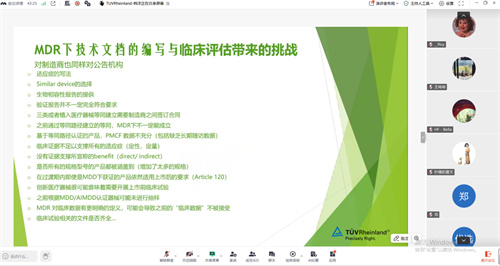
另一方面,MDR法规明确要求企业提供充分、科学的临床证据作为产品安全与性能的支持。临床评价报告必须做到全面、客观,不仅要呈现有利数据,也应如实反映不利证据;其评价的深度与广度,必须与器械的技术特性、风险分类、预期用途、风险水平以及制造商所提出的宣称相匹配。在实践中,临床数据的准备往往存在完整度不足、关键概念混淆、评估人员专业资质不符、文献检索策略不当等问题。
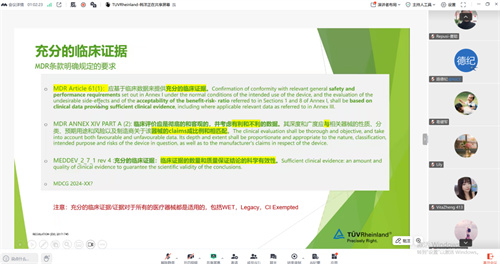
因此,临床评估工作不仅需要严格遵循MDR相关法规与指导文件,确保逻辑链条严谨清晰,还应符合医学常识与临床实践,并始终贯穿风险管理的理念,形成闭环管理。

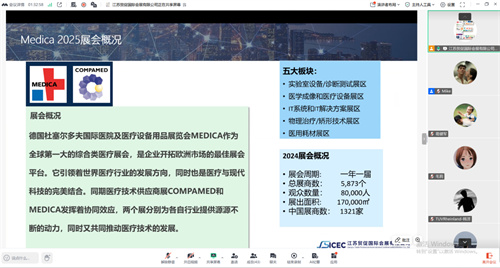
面对当前MDR注册周期长、费用高企的行业现状,我们旨在通过分享专业路径与优质资源,帮助企业规避常见陷阱,优化内部流程,从而显著节约时间与金钱成本,实现更高效、更经济的合规目标。与此同时,作为全球医疗行业风向标的德国杜塞尔多夫国际医院及医疗设备用品展览会MEIDCA即将于下月隆重揭幕,本次会议也希望能为企业提前铺路、占得先机,助力企业提前规划参展策略,最大化参展价值与商务成果。
我们始终致力于为外贸企业提供专业的培训和资源支持,持续为企业传递最新的市场动态和政策准入解读,期待本次内容能为企业带来切实帮助。未来,我们将继续举办更多高质量的分享活动,助力企业把握全球贸易新机遇,实现外贸新突破。欢迎大家持续关注,获取更多前沿资讯与行业信息。